Dawn of Bioinformatics Limited, Bangladesh
1st Bioinformatics Company in our Country

44+
Bioinformatics Services
30+
Training & Courses
100%
Data Security
32+
Team Members
24/7
Dedicated Support
"We are committed to driving innovation in bioinformatics, empowering researchers and industries to transform challenges into breakthroughs. Together, we are not just advancing science in Bangladesh but making a global impact."

Dawn of Bioinformatics Limited Bangladesh has evolved beyond a traditional tech company to become a pioneering, full-scale Bioinformatics Ecosystem. We are a dynamic synergy of advanced computational services, comprehensive scientific education, and groundbreaking research. By bridging the gap between artificial intelligence and life sciences, we provide end-to-end solutions that empower researchers, medical professionals, and students to unlock the mysteries of biology and drive innovation.


About Dawn of Bioinformatics Ecosystem
Our ecosystem is the core of our operations, functioning through three highly specialized and interconnected divisions.


DBS (Dawn Bioinformatics Services)
Acting as the core service engine, DBS offers over 44 specialized services. Our General division provides Genomics, Proteomics, MD Simulation, CADD, and NGS. The Clinical sector focuses on molecular diagnostics and cancer identification. We also provide proprietary Web Application tools for Molecular Docking.

IBAI (Institute of Bioinformatics and Artificial Intelligence)
The ultimate learning hub and permanent solution for bioinformatics education. We cater to all skill levels through beginner short courses, intensive project-based training, 6-month tiered Internship Programs (Levels 1-3), and vital thesis support for MSc students lacking lab facilities.

DawniLab
Our premier R&D laboratory tackling real-world biological challenges using AI. DawniLab developed BMPPD (around 700 plants) and identified potential therapeutic compounds for liver cancer. With over 30 ongoing high-impact projects, it is the heartbeat of our innovation.


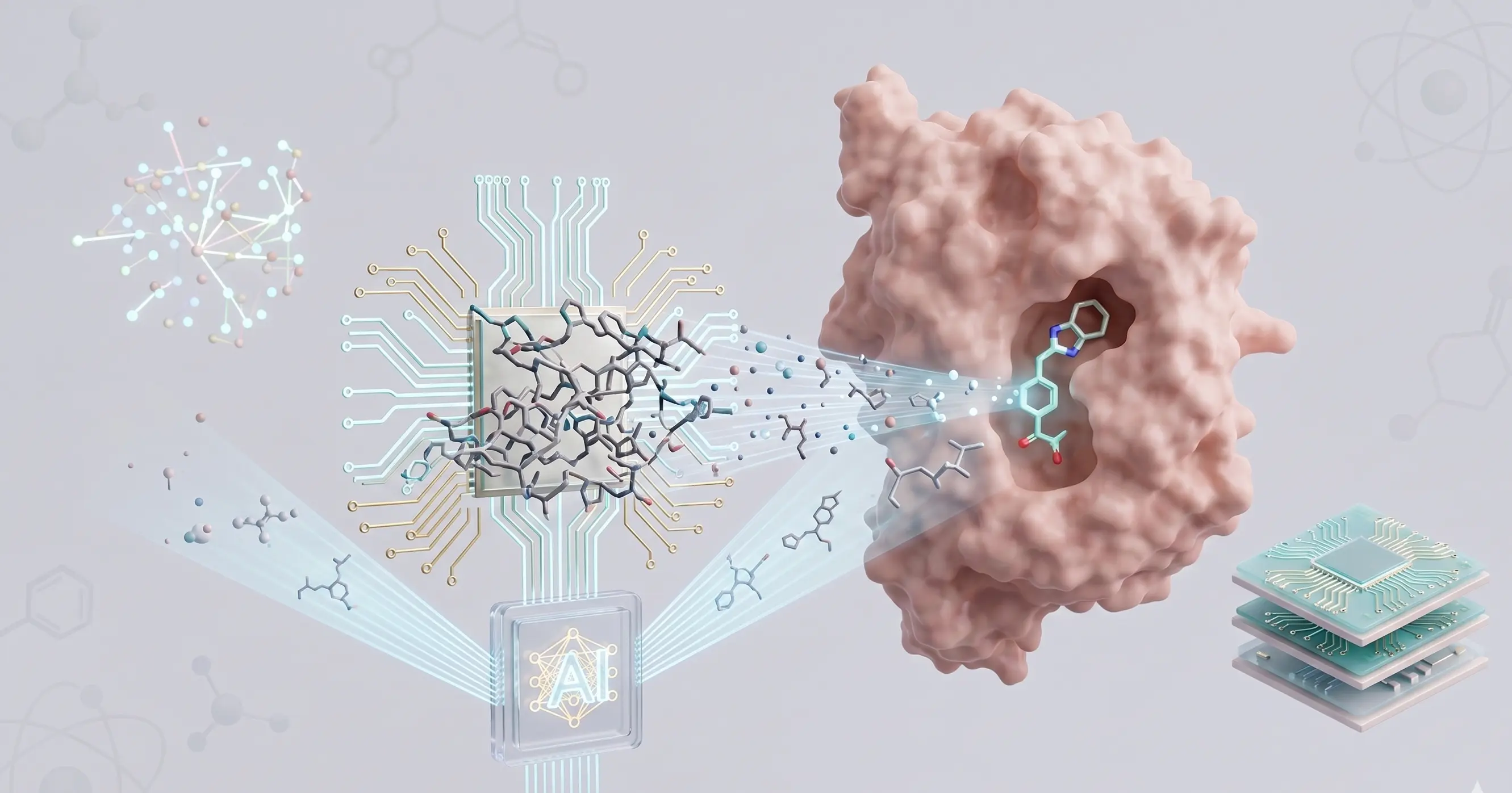
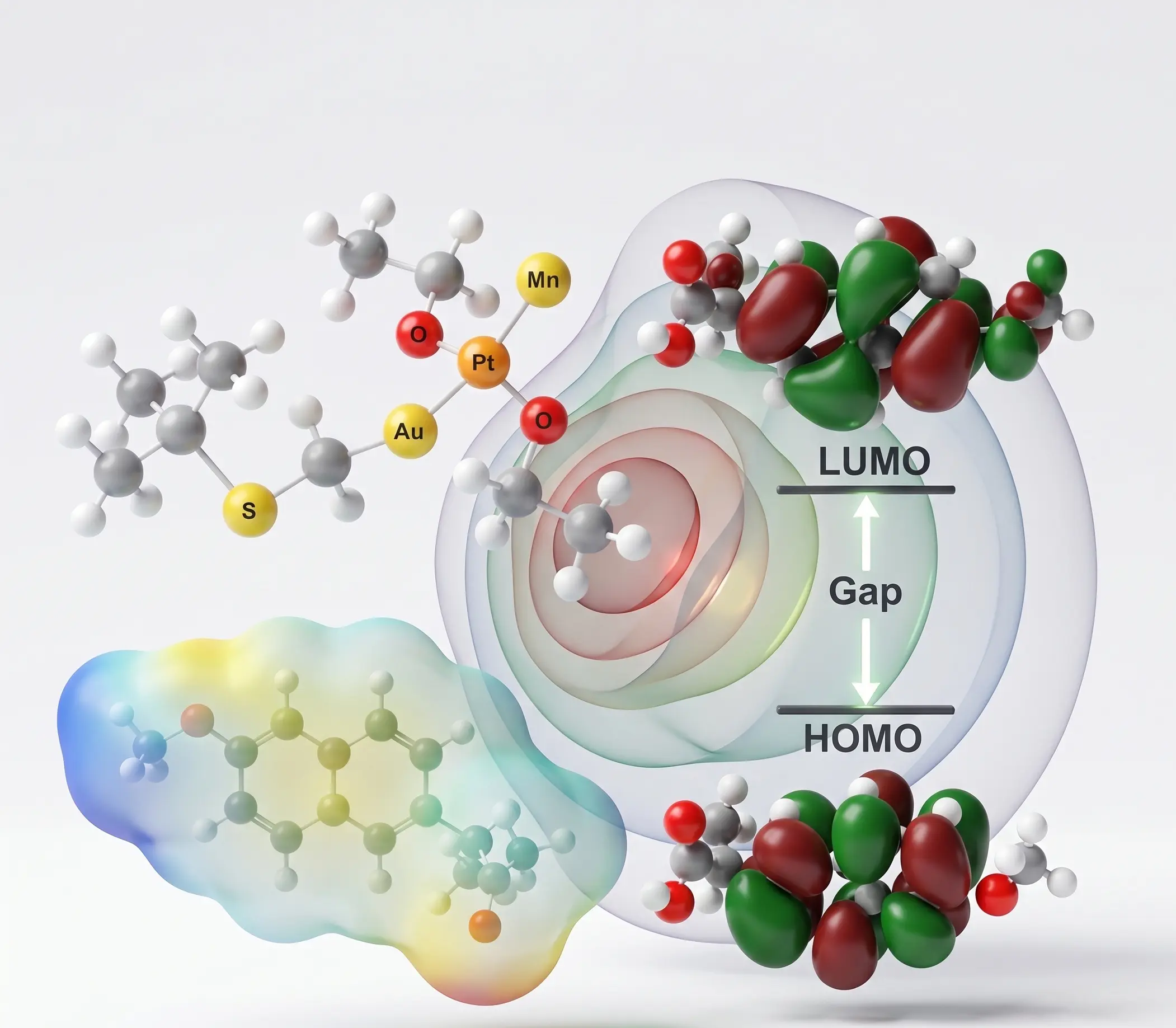









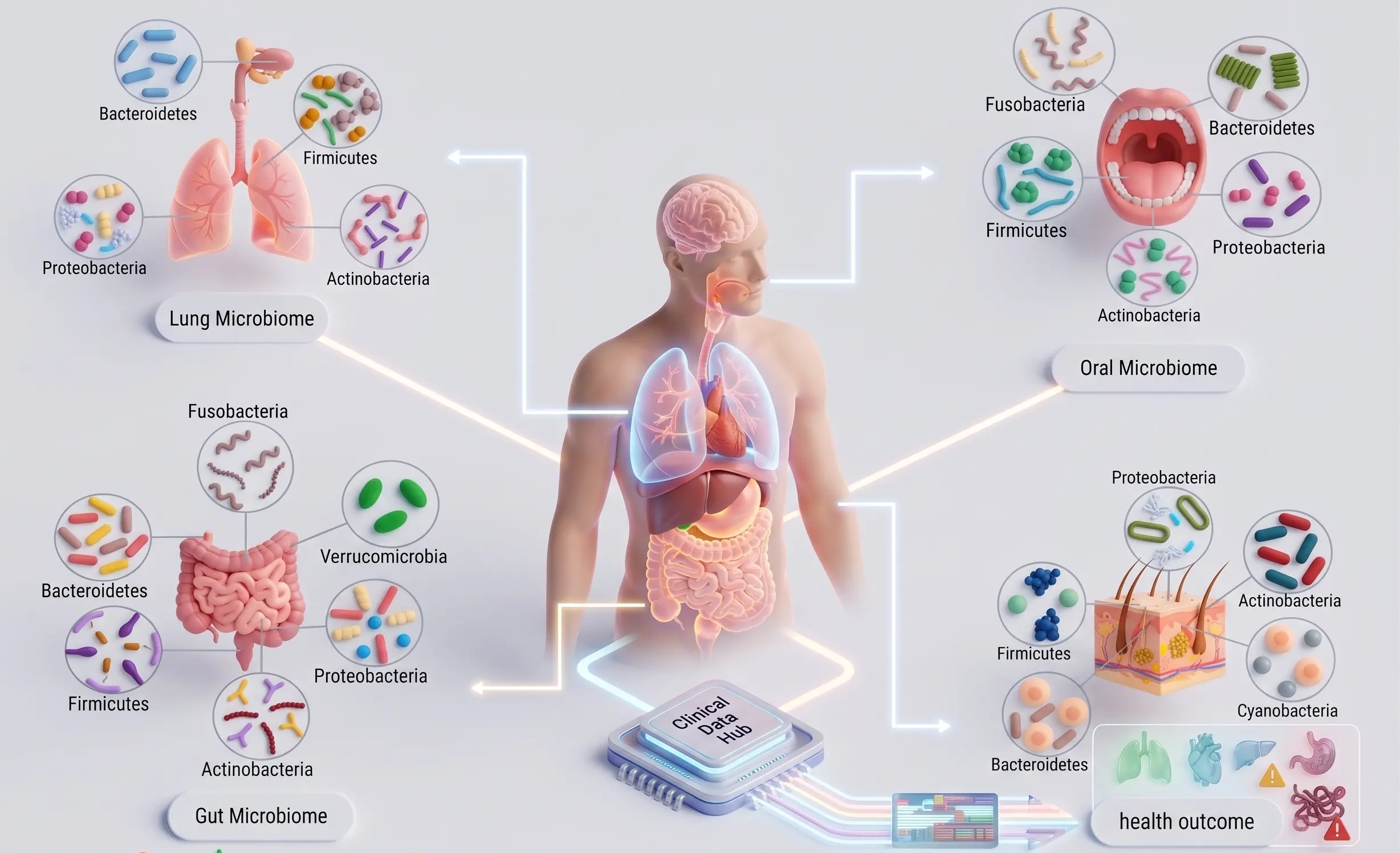






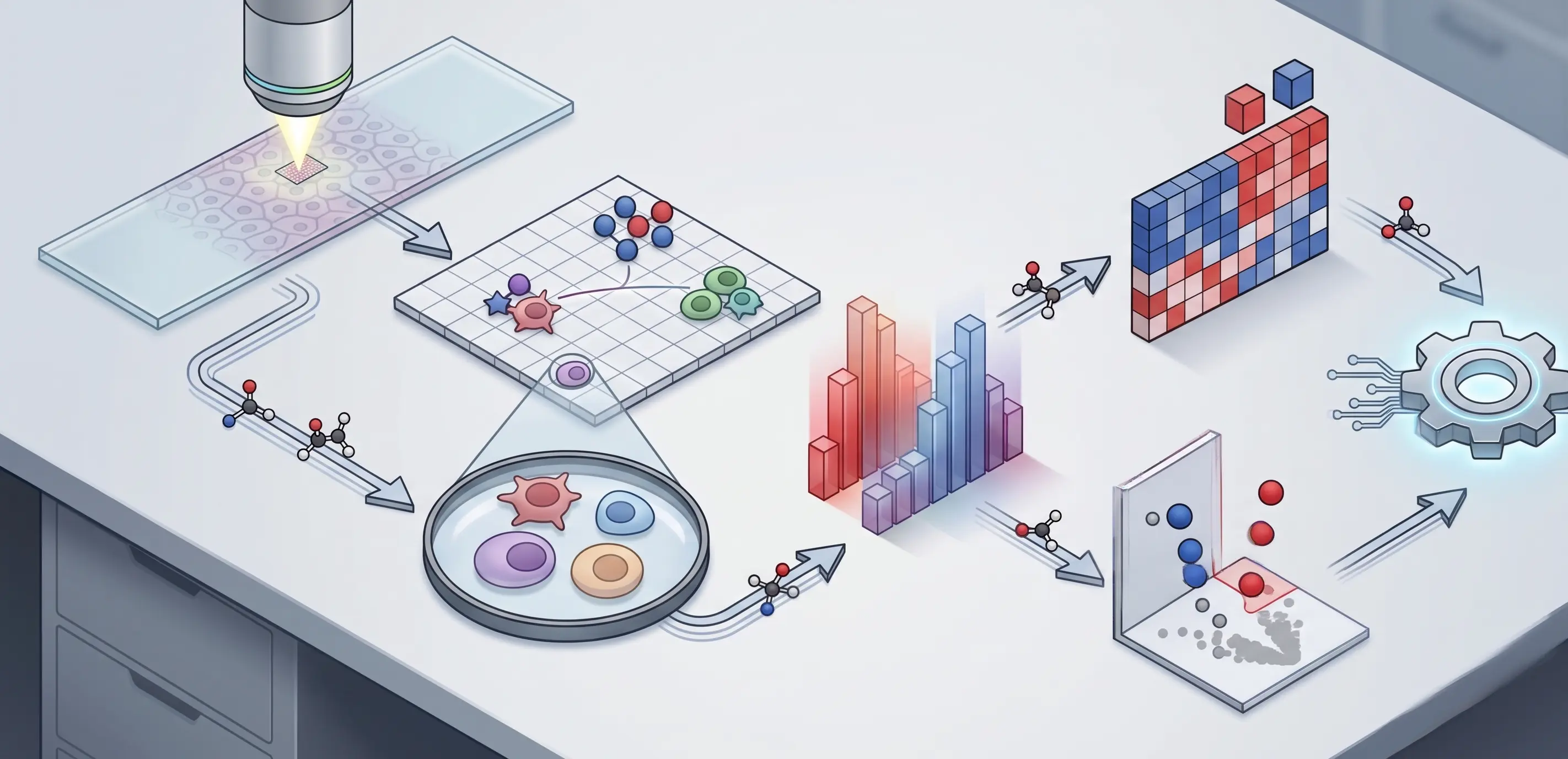



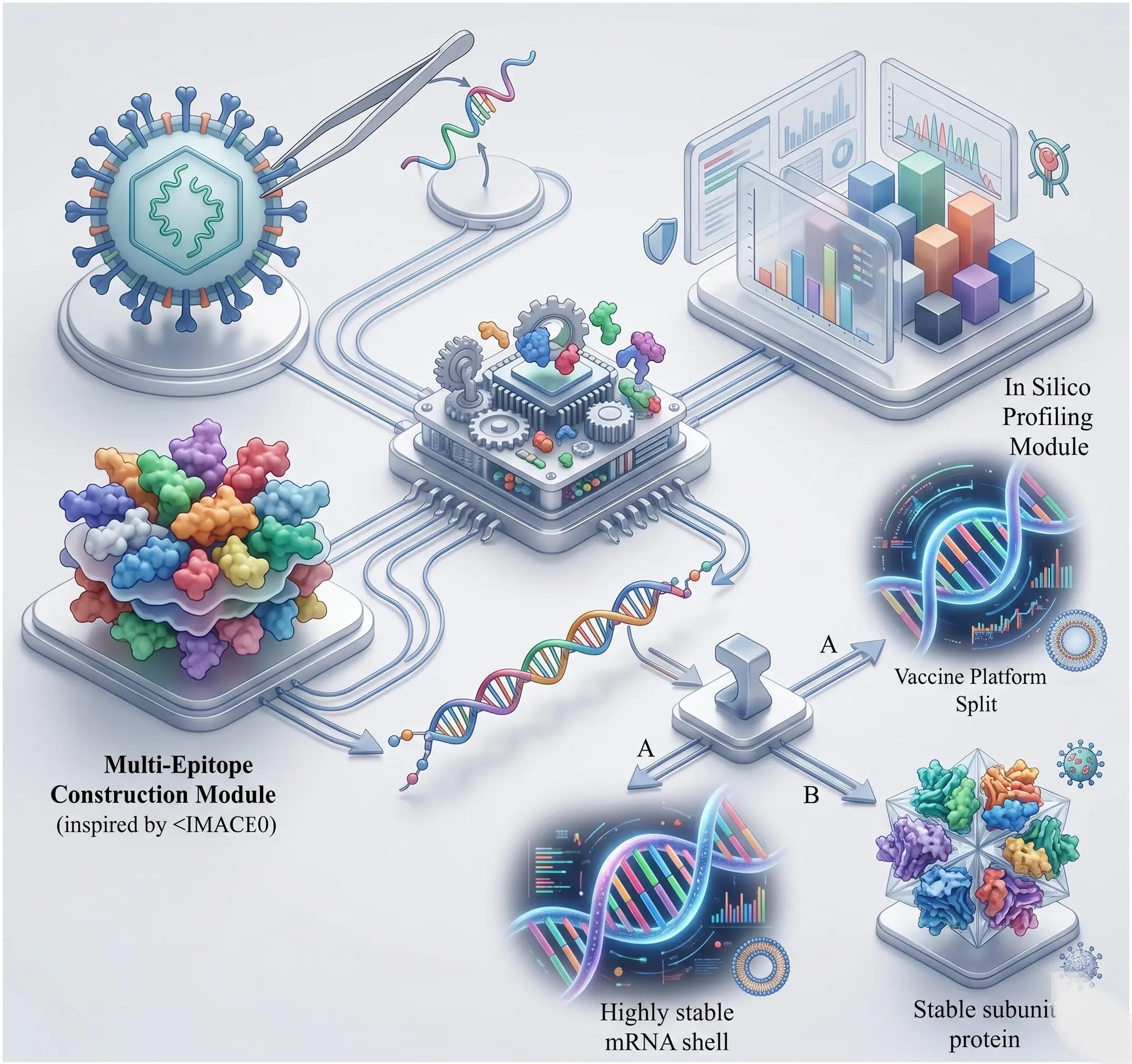








Clinical Bioinformatics
Services

Bridging the gap between complex research and global accessibility.
We host proprietary, cloud-based tools directly on our website. Researchers and students can currently run Molecular Docking, Protein-Protein MD Simulation, and Ligand-Protein MD Simulation seamlessly online, completely eliminating the need for heavy local computing infrastructure.

































BMPPD
Bangladeshi Medicinal Plant Phytochemicals Database
The first curated phytochemical database of medicinal plants in Bangladesh, developed through two years of dedicated research at Dawnilab. It embodies our vision of translating scientific knowledge into practical innovations, bridging the gap between research and application.
Latest Research

In silico evaluation of Ocimum sanctum phytochemicals for diabetic foot ulcer therapy through docking, ADMET, DFT, and molecular dynamics
This study explores the potential of phytocompounds derived from Ocimum sanctum (Holy Basil) to accelerate the healing of diabetic foot ulcers (DFUs). The research targets MMP-9, an enzyme found in elevated levels in DFUs that delays wound repair. Through computational analysis—including molecular docking, ADME toxicity screening, and molecular dynamics simulations—the study identifies apigenin as a superior, stable, and drug-like inhibitor compared to the control drug, (R)-ND-336
Read Paper
Cheminformatics-based analysis identified Novel compounds from Nelumbo nucifera as potential inhibitors targeting PI3k/Akt/mTOR Pathway of HR+/HER2- subtype for Breast Cancer
The present study investigates the in-silico identification of novel phytochemical inhibitors from N. nucifera molecularly targeting the PI3K/Akt/mTOR signalling pathway as a breast cancer chemotherapeutic agent, especially to treat PIK3CA mutant tumours. Despite huge advancements, including the application of various complex intracellular signalling pathways in oncogenesis, breast cancer remains a menace, causing significant morbidity and mortality worldwide.
Read Paper
Computational analysis of miRNA mediated KDR gene regulation and natural VEGFR2 inhibitors from Persicaria hydropiper in hepatocellular carcinoma
Liver cancer is one of the deadliest cancers worldwide. There is a growing need for natural therapeutic options due to the rising global morbidity incidence of liver cancer. Due to its crucial function in angiogenesis, vascular endothelial growth factor (VEGF) signaling is considered as an ideal target for therapeutic intervention. In this in silico study, we have screened 16 microRNAs (miRNAs) that target the KDR gene, which encode VEGFR-2, and 113 natural compounds derived from Persicaria hydropiper for their anti-angiogenic properties.
Read Paper
Licoisoflavone B and glabridin from Glycyrrhiza glabra as potent nucleoprotein antagonists of Lassa virus: insights from molecular docking, dynamics simulation, PCA, and DFT studies
Lassa virus, a deadly pathogen in West and Central Africa with no available vaccine and limited treatment options, urgently requires new therapies. This study screened 69 phytochemicals from Glycyrrhiza glabra and identified licoisoflavone B and glabridin as promising inhibitors of the viral nucleoprotein, crucial for replication and immune evasion. Both compounds showed strong binding affinities, favorable ADMET profiles, and structural stability in 100 ns molecular dynamics simulations, further supported by principal component and density functional theory analyses. These findings highlight licoisoflavone B and glabridin as potential anti-Lassa virus drug candidates, warranting further experimental validation.
Read Paper
In silico identification of promising PD-L1 inhibitors from selected indian medicinal plants for treatment of triple negative breast cancer
Triple-negative breast cancer (TNBC) is a highly aggressive subtype with limited therapeutic targets and poor survival outcomes. Given the limitations of antibody-based PD-1/PD-L1 therapies, this study explored small-molecule phytochemicals as potential PD-L1 inhibitors. A virtual screening of 953 compounds from eleven Indian medicinal plants identified 4-hydroxychalcone (from Glycyrrhiza glabra) and flavylium (from Catharanthus roseus) as top candidates with strong binding affinities, favorable ADMET properties, and high stability in molecular dynamics simulations. Both compounds showed enhanced gastrointestinal absorption and no predicted toxicity, making them promising leads for novel PD-L1 inhibitor development in TNBC therapy, though further experimental validation is required.
Read Paper
In Silico Identification of Emblica officinalis Compounds Inhibiting Thermolabile Hemolysin from Vibrio alginolyticus in Shrimp
Vibrio alginolyticus is a major pathogen in shrimp aquaculture, and reliance on antibiotics raises concerns about resistance. This study used computational approaches to explore garlic (Allium sativum) compounds as alternatives for inhibiting the virulence protein thermolabile hemolysin. Among 35 tested compounds, protopine, gibberellin A7, and gibberellic acid showed the strongest binding affinities, with gibberellin A7 and gibberellic acid demonstrating favorable pharmacokinetics, low toxicity, and high stability in molecular dynamics simulations. These findings suggest that garlic-derived compounds, particularly gibberellin A7 and gibberellic acid, may serve as promising candidates to combat shrimp vibriosis, pending further in vitro and in vivo validation.
Read Paper
Synthesis, antiviral activity, molecular docking, and molecular dynamics studies of ethoxy phthalimide pyrazole derivatives against Cytomegalovirus and Varicella-Zoster virus: potential consequences and strategies for developing new antiviral treatments
Substituted ethoxy phthalimide pyrazole derivatives (6a–e) were synthesized and evaluated for antiviral potential against CMV and VZV. Docking and in vitro studies showed strong, stable binding to key viral proteins, with higher binding energies than standard drugs Ganciclovir and Acyclovir. ADME/T analysis indicated low toxicity, suggesting these compounds as promising multitarget inhibitors and potential adjuvant therapies for CMV and VZV infections.
Read Paper
Identification of novel inhibitors of high affinity iron permease (FTR1) through implementing pharmacokinetics index to fight against black fungus: An in silico approach
This study focuses on identifying novel inhibitors for the FTR1 protein to combat black fungus infections using pharmacokinetic indices. An in silico approach allows for the virtual screening of compounds, predicting their binding affinity and pharmacological properties, thus speeding up the drug discovery process against this pathogen.
Read Paper
Identification of 1, 2, 4-Triazine and Its Derivatives Against Lanosterol 14-Demethylase (CYP51) Property of Candida albicans: Influence on the Development of New Antifungal Therapeutic Strategies
The focus here is on identifying compounds effective against the Lanosterol 14-demethylase (CYP51) enzyme of Candida albicans, a critical target for antifungal therapy. The study evaluates the potential of 1, 2, 4-triazine derivatives in inhibiting CYP51, which could lead to new strategies in antifungal treatment development.
Read Paper
In silico identification of ethoxy phthalimide pyrazole derivatives as IL-17A and IL-18 targeted gouty arthritis agents
This research identifies inhibitors of BNAP, a target associated with the cytokines IL17A and IL18, which are implicated in the inflammatory process of gouty arthritis. Molecular docking and MD simulations, combined with MM-PBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) calculations, are used to predict the efficacy of these inhibitors, providing insights into their potential as therapeutic agents.
Read Paper
In silico identification of potential inhibitors with higher potency than bumetanide targeting NKCC1: An important ion co-transporter to treat neurological disorders
This research aims to find potent inhibitors targeting the NKCC1 ion cotransporter, surpassing the efficacy of Bumetanide, a known inhibitor. NKCC1 is significant in neurological conditions, and its inhibition could be beneficial for treating such disorders. The study employs in silico methods to identify and evaluate potential inhibitors.
Read Paper![Unlocking SGK1 inhibitor potential of bis-[1-N,7-N, pyrazolo tetraethoxyphthalimido{-4-(3,5-Dimethyl-4-(spiro-3-methylpyazolo)-1,7-dihydro-1H-dipyrazolo[3,4-b;4',3'-e]pyridin-8-yl)}]p-disubstituted phenyl compounds: a computational study](/media/research_papers/2-2.webp)
Unlocking SGK1 inhibitor potential of bis-[1-N,7-N, pyrazolo tetraethoxyphthalimido{-4-(3,5-Dimethyl-4-(spiro-3-methylpyazolo)-1,7-dihydro-1H-dipyrazolo[3,4-b;4',3'-e]pyridin-8-yl)}]p-disubstituted phenyl compounds: a computational study
The study explores the inhibitor potential of specific bis-[1-N,7-N, pyrazolo tetraethoxyphthalimido] compounds against the serum and glucocorticoid-regulated kinase 1 (SGK1). Through computational analyses, these compounds are evaluated for their ability to modulate SGK1 activity, which is implicated in various diseases, offering insights into new therapeutic avenues.
Read Paper
Molecular docking and simulation studies of flavonoid compounds against PBP-2a of methicillin‐resistant Staphylococcusaureus
This article examines the effectiveness of flavonoid compounds against the penicillin-binding protein 2a (PBP-2a) of Methicillin-Resistant Staphylococcus aureus (MRSA). Through molecular docking and simulation studies, the research assesses the binding efficiency of flavonoids to PBP-2a, potentially contributing to the development of new treatments for MRSA infections.
Read PaperBSDS Program
Bioinformatics Skill Development System. Empowering students, fostering leadership, and spreading knowledge across campuses.
On-Campus Seminar
Organize impactful seminars to spread computational biology literacy.
Learn More →Campus Coordinator
Become a recognized leader and coordinate bioinformatics activities at your institution.
Apply Now →Research Talk
Host or participate in cutting-edge research discussions and presentations.
Learn More →
Driven by passion,
united by science.
Our interdisciplinary team brings together bioinformaticians, computational chemists, clinical researchers, and software engineers — all united by a shared mission to advance life sciences through cutting-edge technology and open collaboration.

News & Blogs

Strategic Antiviral Discovery: Unveiling Phytochemical Inhibitors Against SARS-CoV-2 Main Protease ($M^{pro}$)
The global effort to counteract the SARS-CoV-2 virus has shifted toward identifying robust therapeutic agents capable of inhibiting viral replication. A peer-reviewed study published in the Journal of Biomolecular Structure and Dynamics, titled "Exploration of potential inhibitors against the main protease ($M^{pro}$) of SARS-CoV-2 from Bangladeshi medicinal plants: an integrated in silico study," highlights the therapeutic potential of regional biodiversity in managing viral outbreaks. Led by Sk. Faisal Ahmed and his research colleagues, this study utilizes an advanced computational pipeline to identify high-affinity lead compounds from indigenous medicinal flora that can effectively neutralize the virus's primary replication machinery. The Significance of the Main Protease ($M^{pro}$) The Main Protease ($M^{pro}$), also known as $3CL^{pro}$, is a critical enzyme for the maturation of the SARS-CoV-2 virus. Since it plays a central role in processing the polyproteins translated from viral RNA, inhibiting $M^{pro}$ effectively halts the viral life cycle. Because this enzyme has no close human homolog, it serves as an ideal target for drug design with minimal risk of off-target effects. Methodological Rigor: An Integrated In Silico Approach The research team employed a multi-layered screening process to ensure the accuracy and reliability of the identified leads: Molecular Docking: Hundreds of phytochemicals derived from Bangladeshi medicinal plants were screened against the $M^{pro}$ binding pocket to assess their binding orientations and affinities. ADMET Profiling: Potential candidates were filtered based on their Absorption, Distribution, Metabolism, Excretion, and Toxicity profiles to ensure pharmacological viability and safety. Molecular Dynamics (MD) Simulation: 100-nanosecond simulations were conducted to evaluate the structural stability and conformational flexibility of the protein-ligand complexes under physiological conditions. Binding Free Energy (MM-PBSA): The thermodynamic stability of the interactions was further validated using MM-PBSA calculations, confirming the strength of the molecular bonds. Key Breakthroughs The study identified several phytochemicals—including those from prominent local species—that exhibited binding affinities superior to many conventional antiviral drugs. These compounds demonstrated exceptional stability within the catalytic site of $M^{pro}$, suggesting they could serve as effective scaffolds for the development of novel, natural-source antiviral therapies. Conclusion: Nature as a Blueprint for Resilience This research underscores the critical importance of documenting and digitizing botanical chemical spaces. By leveraging bioinformatics to validate traditional medicinal knowledge, this study provides a clear roadmap for the rapid development of sustainable and accessible antiviral treatments. As viral variants continue to emerge, such in silico frameworks will remain indispensable in our global health security infrastructure.
Read More
Revolutionizing Structural Bioinformatics: Deep Learning Cracks Complex Multi-Domain Protein Assemblies
Deciphering the 3D architecture of proteins is fundamental to biology and medicine, yet a significant challenge has long persisted: predicting how multiple, large protein domains assemble into functional, complex molecules. A groundbreaking study published in Scientific Reports (2025) has introduced a sophisticated deep learning architecture that drastically improves our ability to predict the structures of these intricate, multi-domain assemblies directly from sequence data. While systems like AlphaFold revolutionized single-chain prediction, predicting multi-domain assemblies, where disjointed parts must fold and fuse correctly, has remained computationally prohibitive. This new research leverages a novel multi-scale, attention-based deep learning model designed to handle the hierarchical complexity of large protein folds. Beyond Single Folds: The Assembly Challenge Protein domains are distinct, independently folding units within a larger protein chain. Conventional AI models often struggle to manage the dynamic spatial relationships between these domains, especially when linked by flexible regions. The Scientific Reports article introduces an 'assembly-aware' transformer model that treats domain folding and inter-domain orientation as a unified computational task. Key Innovations and Architectural Breakthroughs The research team's model operates on two critical views: Sequence-to-Local View: A refined evolutionary transformer predicts local domain structures with high accuracy. Domain-to-Global Fusion View: A specialized graph neural network models the interaction landscape between domains, effectively assembling them like a complex jigsaw puzzle while predicting optimal conformation. By fusing these views, the model achieved unprecedented accuracy on a benchmark of previously computationally resistant multi-domain structures, surpassing conventional fusion methods by a significant margin. Implications for Drug Design and Biological Understanding This advance holds profound implications for rational drug design. Many therapeutic targets are multi-domain proteins; accurately predicting their full structure, including flexible linkers, is essential for designing effective inhibitors or modulators. Furthermore, this tool will accelerate our understanding of vital cellular processes controlled by large macromolecular complexes, from cell signaling to DNA replication. Conclusion: The Age of Holistic Protein Prediction This publication marks a pivotal step toward the holistic prediction of cellular components. Bioinformatics is transitioning from simply mapping sequence to structure, to mapping sequence to complex, dynamic, functional systems. The Scientific Reports study confirms that AI, tailored for hierarchical biological data, will continue to unlock the most fundamental secrets of life's machinery.
Read More
New Frontiers in Neuroinflammation: Uncovering a Novel MicroRNA-Targeted Pathway
Neuroinflammation is a complex and highly regulated process linked to numerous neurodegenerative diseases. While much attention has been focused on protein-coding genes, recent research from Qi Cong, Wenbo Wang, and colleagues, published in Biochemical and Biophysical Research Communications (2025), has identified a critical non-coding RNA that could offer a new therapeutic strategy. The study, titled "MicroRNA-148a-3p suppresses neuroinflammation by targeting the TRAF6/NF-κB pathway," elucidates how a small regulatory molecule, miR-148a-3p, plays a pivotal role in mitigating microglial activation—a hallmark of brain inflammation. Bridging the Gap: MicroRNAs as Key Regulators MicroRNAs (miRNAs) are short, functional RNA molecules that do not code for proteins but instead regulate gene expression. In the context of the brain, dysregulation of miRNAs has been implicated in conditions such as Alzheimer’s and Parkinson’s diseases. This new research provides mechanistic insights into how miR-148a-3p functions to dampen the overactive inflammatory response. Key Insights: A Double-Edged Sword for Inflammatory Signaling The authors utilized a model of microglial cell activation to simulate neuroinflammation. Their findings demonstrate that: Selective Suppression: miR-148a-3p is significantly downregulated in activated, pro-inflammatory microglia. Targeting TRAF6: The study identifies TRAF6 (TNF receptor-associated factor 6) as a direct and functionally relevant target of miR-148a-3p. By binding to TRAF6, the miRNA inhibits its expression. Inhibiting NF-κB: The consequence of this interaction is the suppression of the downstream NF-κB signaling pathway, which is a master regulator of inflammatory gene expression. The final result is a marked reduction in the production of pro-inflammatory cytokines. Implications for Future Therapy The potential significance of these findings cannot be overstated. By acting as a molecular brake on neuroinflammation, miR-148a-3p could serve as both a diagnostic biomarker and a therapeutic target. Developing methods to boost its expression in affected areas of the brain may open new avenues for treating chronic neurodegenerative conditions. This research reinforces the growing understanding that the seemingly "junk" non-coding regions of the genome hold the keys to complex regulatory networks. Bioinformatics and molecular biology continue to merge, revealing elegant mechanisms of control in both health and disease.
Read More
Natural Breakthrough: Ocimum sanctum Phytochemicals as a Therapeutic Frontier for Diabetic Foot Ulcers
Managing Diabetic Foot Ulcers (DFUs) remains one of the most formidable challenges in modern endocrinology, often leading to severe complications including lower extremity amputations. A groundbreaking study recently published in Scientific Reports (2025), titled "In silico evaluation of Ocimum sanctum phytochemicals for diabetic foot ulcer therapy through docking, ADMET, DFT, and molecular dynamics," explores a promising natural alternative to conventional treatments. Led by Sk. Faisal Ahmed and his research team, the study utilizes advanced computational bioinformatics to identify potent inhibitors of MMP-9—an enzyme whose elevated levels are directly linked to delayed wound healing in diabetic patients. Targeting the MMP-9 Barrier In chronic diabetic wounds, the over-expression of MMP-9 disrupts the delicate balance of the extracellular matrix (ECM), hindering the migration of keratinocytes and preventing effective re-epithelialization. This research systematically screened phytochemicals from Ocimum sanctum (Holy Basil) against the known inhibitor (R)-ND-336 to find more effective and safer therapeutic candidates. Key Findings: The Power of Apigenin Through a rigorous multi-stage in silico pipeline—including molecular docking, ADMET profiling, and Density Functional Theory (DFT)—the team identified several high-affinity compounds: Top Candidates: Cianidanol, Luteolin, Rosmarinic acid, Apigenin, and Quercetin showed binding affinities ranging from -9.1 to -9.8 kcal/mol. Stability & Safety: Apigenin emerged as the standout candidate. Molecular dynamics (MD) simulations confirmed its exceptional stability, robust receptor binding, and favorable drug-like properties without hepatotoxicity or neurotoxicity. Clinical Significance The study underscores the potential of plant-derived compounds to restore ECM homeostasis. By specifically inhibiting MMP-9, apigenin could potentially accelerate the "Design-Build-Test-Learn" cycle for new DFU treatments, offering a natural, effective path toward wound recovery and reducing the 5-year mortality risk associated with diabetic complications. Conclusion As bioinformatics continues to bridge the gap between traditional medicine and modern pharmacology, the discovery of apigenin’s role as an MMP-9 inhibitor marks a significant milestone. This research not only validates the therapeutic potential of Ocimum sanctum but also provides a robust computational framework for future drug development in the fight against diabetes-related morbidity.
Read More
The Dawn of Autonomous Science: The Rise of AI Agents in Biological Research
In the rapidly evolving landscape of bioinformatics, the transition from static computational tools to dynamic, autonomous systems marks a pivotal shift in how we approach biological inquiry. A comprehensive new survey published in Briefings in Bioinformatics (2026), titled "Artificial Intelligence agents for biological research," provides a systematic exploration into this new frontier. This research highlights how AI agents—autonomous entities capable of reasoning, planning, and iterative learning—are redefining traditional workflows across clinical analytics, molecular design, and multi-omics integration. Beyond Predictive Modeling: The Agentic Shift For decades, bioinformatics has relied on specialized models designed for specific tasks, such as protein folding or genomic sequence alignment. While powerful, these models often operate in isolation. The authors, Cong Qi, Wenbo Wang, and their colleagues, argue that the next generation of biological research lies in AI Agents. Unlike traditional AI, these agents possess the capability to perceive complex biological environments, make informed decisions, and execute multi-step scientific protocols with minimal human intervention. The 5D Taxonomy: A New Framework for Discovery The survey introduces a robust "5D" taxonomy to categorize the current state of biological AI research: Task Domains: Mapping agents to specific biological challenges, from drug discovery to knowledge synthesis. Architectural Paradigms: Analyzing the underlying structures, such as Large Language Model (LLM)-based reasoning engines. Evaluation Strategies: Establishing rigorous benchmarks for agent reliability and scientific accuracy. Interaction Modes: How these agents collaborate with human scientists and other digital tools. Resource Integration: The seamless connection of agents with biological databases and wet-lab automation. Future Implications for Drug Discovery and Omics One of the most exciting prospects discussed is the role of AI agents in molecular and drug design. By autonomously navigating vast chemical spaces and simulating drug-target interactions, these agents can significantly accelerate the "Design-Build-Test-Learn" cycle. Furthermore, in multi-omics analysis, agents can synthesize disparate data types—genomic, transcriptomic, and proteomic—to reveal holistic biological insights that were previously obscured by data fragmentation. Conclusion: A Collaborative Future As we stand on the brink of this "Agentic Revolution," the integration of AI into biological research is no longer just about faster processing; it is about smarter, more autonomous discovery. While challenges in scalability and reliability remain, the roadmap provided by this survey serves as a cornerstone for researchers aiming to build the next generation of digital scientists.
Read More
লিভার ক্যান্সারের চিকিৎসায় নতুন দিগন্ত: বাংলাদেশি বায়োইনফরমেটিক্সের যুগান্তকারী পদক্ষেপ
লিভার ক্যান্সারের চিকিৎসায় নতুন দিগন্ত: বাংলাদেশি বায়োইনফরমেটিক্সের যুগান্তকারী পদক্ষেপ লিভার ক্যান্সারের মতো মরণব্যাধির চিকিৎসায় নতুন আশার আলো দেখাচ্ছেন বাংলাদেশের একদল তরুণ গবেষক। তাদের এই উদ্ভাবনী গবেষণার মূলে রয়েছেন বায়োইনফরমেটিক্স এবং মলিকিউলার ডায়নামিক্স সিমুলেশন । ফয়সাল আহমেদ ও তার দল দেশীয় প্রযুক্তির মাধ্যমে লিভার ক্যান্সারের চিকিৎসায় একটি সাশ্রয়ী ও কার্যকর পদ্ধতির উদ্ভাবনের পথে এগিয়ে যাচ্ছেন, যা বাংলাদেশের স্বাস্থ্যখাতে এক বৈপ্লবিক পরিবর্তন আনতে পারে। গবেষণার মূল উদ্ভাবন টার্গেটেড থেরাপি: ফারহিম আহমেদের গবেষণা দল মূলত লিভার ক্যান্সারের কোষের বিরুদ্ধে কাজ করে এমন প্রাকৃতিক উপাদান বা ওষুধের কার্যকারিতা নিয়ে কাজ করছে। তারা এমন কিছু অণু চিহ্নিত করেছেন যা ক্যান্সার কোষের বৃদ্ধি রোধ করতে পারে। কম্পিউটেশনাল মডেলিং: এই গবেষকরা অত্যাধুনিক কম্পিউটার মডেলিং এবং বায়োইনফরমেটিক্স টুলস ব্যবহার করে নিশ্চিত করেছেন যে নির্বাচিত অণুগুলো মানবদেহে কী ধরনের প্রভাব ফেলবে এবং কীভাবে সবচেয়ে কার্যকরভাবে ক্যান্সার কোষকে লক্ষ্যবস্তু করবে। এই পদ্ধতি চিকিৎসার পার্শ্বপ্রতিক্রিয়া কমাতে সাহায্য করবে। সাশ্রয়ী সমাধান: এই উদ্ভাবন স্থানীয় ও সহজলভ্য উপাদান ব্যবহার করে চিকিৎসা পদ্ধতির ব্যয় উল্লেখযোগ্যভাবে হ্রাস করতে পারে, যা উন্নত চিকিৎসা অনেকের জন্য সহজলভ্য করবে।
Read More
আমার চোখে স্যার Erwin Schrödinger এবং MD Simulation
গত ৮ বছরেরও বেশি সময় বায়োইনফরমেটিক্সে কাজের সুবাধে, এই ফিল্ডের বিভিন্ন সেক্টরে আমার গবেষণা করা হয়েছে। আর এই গবেষণার পথচলায় যখনই নতুন কোন ড্রাগ বা ভ্যাকসিনের কাজ করেছি, তখনই প্রায় নিয়মিতই আমাকে Molecular Dynamics Simulation রান দিতে হয়েছে— যেটি মূলত ছিল প্রোটিন-লিগ্যান্ড বা প্রোটিন-প্রোটিন ইন্টার‌্যাকশনের গভীর রহস্য উন্মোচনে এটি এক অনন্য হাতিয়ার। আজ আমার এই লেখায় আমি এমন একজন বিজ্ঞানীর কথা বলবো, যিনি আজকের এই আধুনিক MD simulation এর তাত্ত্বিক ভিত্তি গড়ে দিয়েছিলেন (Erwin Schrödinger)। মূলত Erwin Schrödinger ছিলেন একজন অস্ট্রিয়ান পদার্থবিজ্ঞানী। যিনি ১৯৩৩ সালে তাঁর বৈপ্লবিক "Schrödinger Equation"-এর জন্য Nobel Prize পেয়েছিলেন , যা তখন কোয়ান্টাম মেকানিক্স-এর ভিত্তি নির্মাণে অসাধারণ ভূমিকা রেখেছিল। যদিও তিনি সরাসরি বায়োইনফরমেটিক্স বা ড্রাগ ডিজাইনের সাথে কাজ করেননি, কিন্তু তাঁর গবেষণার ফল আজও এই ফিল্ডে আমরা ব্যবহার করছি জীবনরহস্যের নতুন কিছু উন্মোচনে। যদি আমি MD Simulation-নিয়ে একটু বলি, এটি আসলে একটা মলিকিউল কীভাবে সময়ের সাথে সাথে তার আচরণ পরিবর্তন করে, সেটাকে বোঝার একটি পক্রিয়া। আমাদের ভাষায় আমরা এটিকে বলি "atomistic motion over time"। আর এই গতির পিছনে যে গাণিতিক কাঠামো কাজ করে, তার ভিত্তি যে বিষয়টির উপর দাঁড়িয়ে আছে সেটি হল Schrödinger Equation। অবাক করা বিষয় হল এই সমীকরণটা কিন্তু কেবল পদার্থবিজ্ঞানের জন্য নয়—জীববিজ্ঞানের অণু-পর্যায়ের সমস্যাগুলো বোঝার জন্যও অপরিহার্য। আমরা যখন একটা প্রোটিন আর একটা সম্ভাব্য ড্রাগ মলিকিউলের ইন্টার‌অ্যাকশন দেখি—তখন আমরা মূলত তাদের কণাগুলোর quantum-level behavior-এর একটা simplification-এর উপর ভিত্তি করে সিদ্ধান্ত নেই। এবং এই simplification-এ Schrödinger-এর কাজই হলো ভিত্তিভূমি। আশ্চর্যের বিষয় হলো—আজকে আমরা যারা computational drug design-এ কাজ করি, তারা অনেকেই “Schrödinger Suite” নামক এক বিশাল সফটওয়্যার প্যাকেজ ব্যবহার করি—যা protein-ligand docking, MD simulation, binding energy calculation থেকে শুরু করে নানা ধরণের বিশ্লেষণ করতে সক্ষম। অথচ আমরা অনেকেই জানি না এর ইতিহাস কি, কিংবা কোন ইকুয়েশনে এটি কাজ করে। সত্যি বলতে এই সফটওয়্যারের নাম শুধু শ্রদ্ধা স্বরূপ নয়, বরং এই নামের মাধ্যমে আমরা এক বৈজ্ঞানিক উত্তরাধিকারের ধারক-ভবিষ্যৎ বাহক হয়ে উঠি। অবশেষে এটা বলাই বাহুল্য Schrödinger Equation ছাড়া হয়তো কখনোই MD Simulation আজকের মতো কার্যকরী হতে পারত না। আর MD Simulation ছাড়া বর্তমান যুগে ওষুধ আবিষ্কারের যে যুগান্তকারী পরিবর্তন সাধন হয়েছে সেটিও হত না। একজন বায়োইনফরমেটিসিয়ান হিসেবে, যখন আমি GROMACS বা Desmond দিয়ে simulation রান করেছি তখনি এই মানুষটির কথা স্মরণে এসেছে, এবং তার একটি বিখ্যাত উক্তি মনের মধ্যে বরাবরি একটু উকি দিয়ে গেছে: "What is life?" হয়তো এই প্রশ্নের উত্তর খুঁজতেই আজও আমি বায়োইনফরমেটিক্সে পথে !
Read More
আমার চোখে Margaret Oakley Dayhoff এবং Dawn of Bioinformatics
আমরা যখন বায়োইনফরমেটিক্স নিয়ে কাজ করি, কম্পিউটারের স্ক্রিনে বসে বিভিন্ন Protein Sequening Data Analysis করি কিংবা নতুন কোন Protein Structure এর পরিবর্তন নিয়ে চিন্তিত হই , তখন কি কখনো মনে হয় এই ফিল্ডটার শুরু কোথা থেকে হয়েছিল, কিংবা কে প্রথম ভেবেছিল, কম্পিউটারের কিছু কোডের ভাজে, আমরা আমাদের জীবন রহস্যের কোডগুলোও ভাঙতে পারবো? এই প্রশ্নের উত্তর খুঁজতে গিয়েই আমি একদিন আবিষ্কার করেছিলাম এক অসাধারণ নারী বিজ্ঞানীকে—Margaret Oakley Dayhoff (The Dawn of Bioinformatics) যার নামটি ইতিহাসের পাতায় হয়তো কখনোই ঐভাবে আলোচিত ছিলনা , কিন্তু যেদিন প্রথম protein FASTA ফাইলটা খুলে হাতে নিয়েছিলাম, PAM matrix টি ব্যবহার করে Protein sequence alignment করেছিলাম—সেদিন হয়তো নিজের অজান্তেই হেঁটেছি তাঁর রেখে যাওয়া পথে। আর এরপর যতই বায়োইনফরমেটিক্সের গভীরে যাত্রা শুরু করেছি, ততই জেনেছি এই মানুষটার সম্পর্কে, জেনেছি তাঁর হাত ধরে তৈরি হওয়া প্রথম প্রোটিন সিকোয়েন্স ডাটাবেস, যেটিই ছিল বায়োইনফরমেটিক্সে সেই সময়ের একমাত্র ডাটাবেস, এখানে বলে রাখা ভালো যে সালটা তখন ১৯৬৫ । আর এরপর তার দেয়া PAM Matrix, যেটি এখনও সকলে নিজের অজানতেই ব্যবহার করে চলেছে, PBLAST এর আড়ালে,এছাড়াও তিনি তৈরি করেছিলেন “Atlas of Protein Sequence and Structure” । সবচেয়ে অবাক করার বিষয় হল এই মানুষটি যে সময়ে এই অসাধারণ কাজগুলো করেছে তখন বায়োইনফরমেটিক্সের সূচনাতো ঘটেইনি আর না ঐভাবে ঘটেছিল কম্পিউটারের বিকাশ ! একজন নারী হয়ে তিনি যেভাবে ১৯৬০–৭০ দশকে এই অসাধারণ কাজগুলো করেছিলেন, সেটা শুধু আমার কাছেই নয় বরং সকলের কাছেই অনেক বড় অনুপ্রেরণা। আর আমি যখনই PBLAST রান করি, কিংবা কোন Protein sequence নিয়ে কাজ করি—তখনই মনে হয়, কোনো এক সন্ধ্যায় এই মানুষটি হয়তো ভেবেছিল এই অসম্ভব চিন্তাটা—"Can biology be digitized?" হয়তো সেই ভাবনার উত্তর খুঁজতে গিয়েই শুরু হয়েছিল বায়োইনফরমেটিক্সের গল্পটি (Dawn of Bioinformatics)। আর যেই গল্পের সূচনাকার ছিলেন—Margaret Oakley Dayhoff। আজও যখন কোন তরুণ গবেষক আমার কোন বায়োইনফরমেটিক্স ক্লাসে বসে প্রশ্ন করে, কিভাবে শুরু হল এই বায়োইনফরমেটিক্সের যাত্রা, আমি তখন গর্বের সাথে বলি—এই পথটা এসেছিল একজন স্বপ্নবাজ নারীর হাতে গড়া একটি অসম্ভব স্বপ্ন থেকে। তিনি ছিলেন না কোনো বিজ্ঞান-পুষ্ট নায়ক, না ছিলেন কোনো নোবেলজয়ী। কিন্তু তাঁর উত্তরাধিকার আজ কোটি কোটি মানুষের জীবন রক্ষায় নিয়োজিত, খুঁজে বেড়াতে ব্যস্ত কঠিন সব রোগের সমাধানের কোড । আর আমার নিজের এই বায়োইনফরমেটিক্স যাত্রায় যখনই ক্লান্তি আসে, তখনই এই প্রশ্নটা নিজের মধ্যে জেগে উঠে - "তুমি যেটা করছো, সেটা কি আগামী দিনের কারো জীবন বদলে দিতে পারবে?”
Read MoreJoin Our Growing Ecosystem
Whether you are a researcher looking for technical support, a student eager to learn, or an institution seeking collaboration, there is a place for you at Dawn of Bioinformatics.
Over 100+ people trust us
Leading researchers, students, and biotech executives rely on Dawn of Bioinformatics for precision biological data analysis and breakthrough scientific discovery.
"The team’s expertise in CADD services helped me gain deep insights into ligand-protein interactions for my thesis."

Lamia Islam
Khulna University
"The gene ontology and pathway enrichment analysis provided in the RNAseq service gave me a clear biological perspective on my experimental data."
Jarin Tasnim
Mawlana Bhashani Science and Technology University
"Excellent presentation and the trainers were very responsive to questions."
Tanvir Ahmed
Bangladesh University of Engineering and Technology
"Your cooperative is excellent. Your lecture materials supply its very helpful personally for me."
Mehnaz Tabassum
BRAC University
"Their support in managing complex RNAseq pipelines was outstanding and helped me solve several troubleshooting issues."
Afsana Mim
Jagannath University
"The NGS data analysis service was extremely fruitful; the pipeline was clear and the results were publication-ready."
Sazzad Hossain
Rajshahi University
"Highly impressed with the precision of the MD simulation results and the comprehensive reporting style."
Ahsan Habib
Chittagong University
"The MD simulation service was good."
Aisha Rahman
University of Dhaka
"This session was very fruitful to me to learn NGS data analysis. Vaiya taught us comprehensively."
Nusrat Jahan
Rajshahi University
"I appreciate the effort put into making complex topics simple. Thank you for this opportunity."
Mahir Asif
BRAC University
"Received high-quality metagenomic analysis with very rational and informative interpretations of the microbial community."
Farhana Yasmin
Bangladesh Agricultural University
"The MD simulation analysis provided for my project was exceptionally detailed and accurate."
Kamrul Islam
Jahangirnagar University
"The team’s expertise in CADD services helped me gain deep insights into ligand-protein interactions for my thesis."
Lamia Islam
Khulna University
"The gene ontology and pathway enrichment analysis provided in the RNAseq service gave me a clear biological perspective on my experimental data."
Jarin Tasnim
Mawlana Bhashani Science and Technology University
"Excellent presentation and the trainers were very responsive to questions."
Tanvir Ahmed
Bangladesh University of Engineering and Technology
"Your cooperative is excellent. Your lecture materials supply its very helpful personally for me."
Mehnaz Tabassum
BRAC University
"Their support in managing complex RNAseq pipelines was outstanding and helped me solve several troubleshooting issues."
Afsana Mim
Jagannath University
"The NGS data analysis service was extremely fruitful; the pipeline was clear and the results were publication-ready."
Sazzad Hossain
Rajshahi University
"Highly impressed with the precision of the MD simulation results and the comprehensive reporting style."
Ahsan Habib
Chittagong University
"The MD simulation service was good."
Aisha Rahman
University of Dhaka
"This session was very fruitful to me to learn NGS data analysis. Vaiya taught us comprehensively."
Nusrat Jahan
Rajshahi University
"I appreciate the effort put into making complex topics simple. Thank you for this opportunity."
Mahir Asif
BRAC University
"Received high-quality metagenomic analysis with very rational and informative interpretations of the microbial community."
Farhana Yasmin
Bangladesh Agricultural University
"The MD simulation analysis provided for my project was exceptionally detailed and accurate."
Kamrul Islam
Jahangirnagar University